
SLIDE 1
1
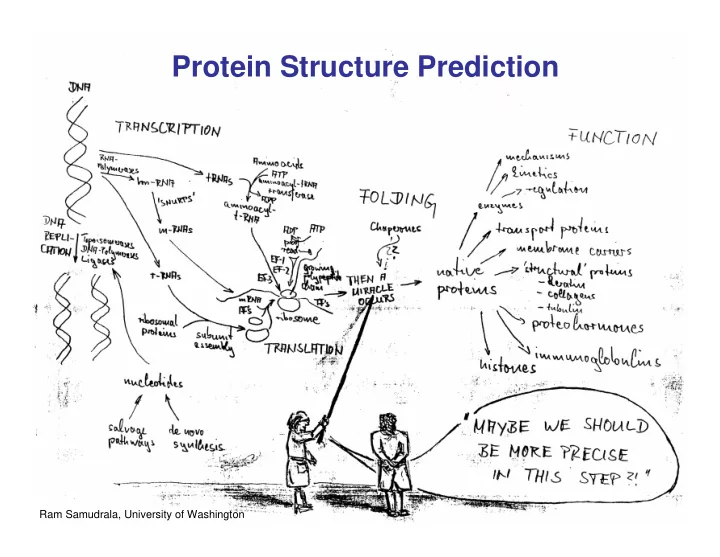
Ram Samudrala, University of Washington
Protein Structure Prediction 1 Ram Samudrala, University of - - PowerPoint PPT Presentation
Protein Structure Prediction 1 Ram Samudrala, University of Washington Rationale for Understanding Protein Structure and Function structure determination Protein sequence structure prediction -large numbers of sequences, including Protein
1
Ram Samudrala, University of Washington
2
Protein sequence
sequences, including whole genomes
Protein function
structure determination structure prediction homology rational mutagenesis biochemical analysis model studies
Protein structure
3
…-L-K-E-G-V-S-K-D-… …-CUA-AAA-GAA-GGU-GUU-AGC-AAG-GUU-…
DNA protein sequence unfolded protein native state
spontaneous self-organization (~1 second) not unique mobile inactive expanded irregular
4
…-L-K-E-G-V-S-K-D-… …-CUA-AAA-GAA-GGU-GUU-AGC-AAG-GUU-…
DNA protein sequence unfolded protein native state
spontaneous self-organisation (~1 second) unique shape precisely ordered stable/functional globular/compact helices and sheets not unique mobile inactive expanded irregular
5
unfolded
Large multi-dimensional space of changing conformations
free energy folding reaction
molten globule J=10-8 s native J=10-3 s ΔG*
*
RT G
*
Δ −
barrier height
6
twenty types of amino acids R H C OH O N H H Cα two amino acids join by forming a peptide bond R Cα H C O N H H N Cα H C O OH R H R Cα H C O N H N Cα H C O R H R Cα H C O N H N Cα H C O R H χ χ χ χ φ φ φ φ ψ ψ ψ ψ each residue in the amino acid main chain has two degrees of freedom (φ and ψ) the amino acid side chains can have up to four degrees of freedom (χ1-4)
7
β α L φ 0 0 ψ
+180 +180
many φ,ψ combinations are not possible
α helix β sheet (anti-parallel)
N C N C
β sheet (parallel)
8
Ribonuclease inhibitor (2bnh) Haemoglobin (1hbh) Hemagglutinin (1hgd)
9
Protein sequence
sequences, including whole genomes
Protein function
X-ray crystallography NMR spectroscopy homology rational mutagenesis biochemical analysis model studies
Protein structure
10
Let’s see how this could work…
interactions between them, we could get to the solution of the folding problem So, why is it then so complicated…
ab initio !!!
11
What should we do?
12
Protein sequence
sequences, including whole genomes
Protein function
comparative modeling fold recognition ab initio prediction homology rational mutagenesis biochemical analysis model studies
Protein structure
13
Protein Sequence Database Searching Domain Assignment Multiple Sequence Alignment Homologue in PDB Comparative Modelling Secondary Structure and Disorder Prediction
No Yes
3-D Protein Model Fold Recognition Predicted Fold Sequence-Structure Alignment Ab-initio Structure Prediction
No Yes
modified from http://bioinf.cs.ucl.ac.uk
14
dimensional structures
15
KDHPFGFAVPTKNPDGTMNLMNWECAIP KDPPAGIGAPQDN----QNIMLWNAVIP ** * * * * * * * **
… … scan align
build initial model
construct non-conserved side chains and main chains
refine
16
17
18
19
Recognition of similarity between the target and template Target – protein with unknown structure. Template – protein with known structure. Main difficulty – deciding which template to pick, multiple choices/template structures. Template structure can be found by searching for structures in PDB using sequence-sequence alignment methods.
20
50 100 150 200 50 100
Safe homology modeling zone Twilight zone
Alignment length Sequence identity
21
1. If alignment between target and template is ready, copy the backbone coordinates of those template residues that are aligned. 2. If two aligned residues are the same, copy their side chain coordinates as well.
22
insertion
deletion
Occur mostly between secondary structures, in the loop regions. Loop conformations – difficult to predict. Approaches to loop modeling:
Energy minimization or Monte Carlo.
23
Scan database and search protein fragments with correct number of residues and correct end-to-end distances
24
Side chain conformations – rotamers. In similar proteins - side chains have similar conformations. If % identity is high - side chain conformations can be copied from template to
rotamers and different rotamers are scored with energy functions. Problem: side chain configurations depend on backbone conformation which is predicted, not real
E1 E2 E3
25
vice versa - iterative approach
26
CASP5 assessors, homology modeling category: “We are forced to draw the disappointing conclusion that, similarly to what observed in previous editions of the experiment, no model resulted to be closer to the target structure than the template to any significant extent.” The consensus is not to refine the model, as refinement usually pulls the model away from the native structure!!
27
BC excellent ~ 80% 1.0 Å 2.0 Å alignment side chain short loops longer loops
28
CASP1 poor ~ 50% ~ 3.0 Å > 5.0 Å BC excellent ~ 80% 1.0 Å 2.0 Å alignment side chain short loops longer loops
29
Cα RMSD of 1.0 Å for 198 residues (PID 50%)
30
Cα RMSD of 2.9 Å for 241 residues (PID 33%)
31
Cα RMSD of 4.4 Å for 137 residues (PID 24%)
32
Cα RMSD of 4.9 Å for 348 residues (PID 24%)
33
Cα RMSD of 5.6 Å for 104 residues (PID 12%)
34
CASP4: overall model accuracy ranging from 1 Å to 6 Å for 50-10% sequence identity
**T112/dhso – 4.9 Å (348 residues; 24%) **T92/yeco – 5.6 Å (104 residues; 12%) **T128/sodm – 1.0 Å (198 residues; 50%) **T125/sp18 – 4.4 Å (137 residues; 24%) **T111/eno – 1.7 Å (430 residues; 51%) **T122/trpa – 2.9 Å (241 residues; 33%)
CASP2 fair ~ 75% ~ 1.0 Å ~ 3.0 Å CASP3 fair ~75% ~ 1.0 Å ~ 2.5 Å CASP4 fair ~75% ~ 1.0 Å ~ 2.0 Å CASP1 poor ~ 50% ~ 3.0 Å > 5.0 Å BC excellent ~ 80% 1.0 Å 2.0 Å alignment side chain short loops longer loops
35
experimentally!
from those experimental structures
generate a model.
36
Proteins with known structures Unknown proteins
37
sequence
sequence-sequence alignment methods fail
38
but few folds)
dimensional structures
known structure and the “goodness of fit” is evaluated using a discriminatory function
3.6 Å 5% ID
NK-lysin (1nkl) Bacteriocin T102/as48 (1e68)
39
KDHPFGFAVPTKNPDGTMNLMNWECAIP KDPPAGIGAPQDN----QNIMLWNAVIP ** * * * * * * * **
… … evaluate fit
build initial model
construct non-conserved side chains and main chains
refine
40
41
Target Sequence α & β structure from template structure
Template
42
– target sequence is compared to all structural templates from the database
Requires:
– dynamic programming, Monte Carlo,…
– yields relative score for each alternative alignment
43
A representative set of protein structures extracted from the PDB
1. The resolution of each representative structure should be good; 2. A good X-ray structure has higher priority than an NMR structure; 3. The sequence identity between any two representatives should be no more than 30%, in order to save computing time. Examples:
44
and distance between them.
function does not depend on the distance between two residues.
residues in the template is less than 8Å, these residues make a contact.
45
) , ( ) , ( ; ) , (
1 ,
Trp Ile w Tyr Ala w S a a w S
N j i j i
+ = = ∑
=
Ala Ile Tyr Trp w - calculated from the frequency of amino acid contacts in PDB
ai - amino acid type of target sequence aligned with the position i of the template N - number of contacts
46
Class work: calculate the score for target sequence “ATPIIGGLPY” aligned to the template structure which is defined by the contact matrix.
* * 10 9 * 8 * 7 * 6 * * 5 * 4 * 3 2 * * * 1 10 9 8 7 6 5 4 3 2 1 0.3 L 0.2 0.4 G 0.4 0.2 0.3 I
Y
0.1
P 0.1
0.3 T 0.2
0.5
A L G I Y P T A
=
=
N j i j i a
a w S
1 ,
) , (
47
“frozen approximation”: traceback in the alignment matrix is not possible for interactions between two amino acids, so that:
1 ,
=
N j i j i b
b – amino acid type from template, not from target; now the score of every position does not depend on the alignment elsewhere in the sequence.
48
– Interaction-Frozen Algorithm (A. Godzik et al.) – Monte Carlo Sampling (S.H. Bryant et al.) – Double dynamic programming (D. Jones et al.)
– Branch-and-bound (R.H. Lathrop and T.F. Smith) – PROSPECT-I uses Divide-and-conquer (Y. Xu et al.) – Linear programming by RAPTOR (J. Xu et al.)
49
– e.g. SAMT02 (K. Karplus et al.)
– e.g. PDB-Blast (A. Godzik et al.)
– e.g. PROSPECT-II (Y. Xu et al.)
– e.g. 3DPS (L.A. Kelley et al), SHGU (D. Fischer)
50
>> 3.8 Angstroms
51
Prediction of structure of methylglyoxal synthase based on the template of carabamoyl phosphate synthase
52
1. Predicts secondary structures for target sequence 2. Makes sequence profiles (PSSMs) for each template sequence 3. Uses threading scoring function to find the best matching profile
http://bioinf.cs.ucl.ac.uk/psipred
53
54
Protein Sequence Database Searching Domain Assignment Multiple Sequence Alignment Homologue in PDB Comparative Modelling Secondary Structure Prediction Disorder Prediction
No Yes
3-D Protein Model Fold Recognition Predicted Fold Sequence-Structure Alignment Ab-initio Structure Prediction
No Yes
http://bioinf.cs.ucl.ac.uk
55
56
57
Non-bonded pair
58
2 0)
( b b K U − =
2 0)
( θ θ − = K U
b θ φ All chemical bonds Angle between chemical bonds Preferred conformations for Torsion angles:
(aromatic, …)
59
⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ ⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ − ⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ =
6 12
2 ) ( r R r R r E
ij ij ij LJ
ε
1/r12 1/r6 Rij r Lennard-Jones potential
j i ij j i ij
R R R ε ε ε = + = ; 2
60
r Coulomb potential qi qj
61
ENCAD (Michael Levitt, Stanford) AMBER (Peter Kollman, UCSF; David Case, Scripps) CHARMM (Martin Karplus, Harvard) OPLS (Bill Jorgensen, Yale) MM2/MM3/MM4 (Norman Allinger, U. Georgia) ECEPP (Harold Scheraga, Cornell) GROMOS (Van Gunsteren, ETH, Zurich)
Michael Levitt. The birth of computational structural biology. Nature Structural Biology, 8, 392-393 (2001)
62
63
64
65
66
> 5 Å 20 ns (20 ps) ms – hrs Global protein tumbling (water tumbling) protein folding 1-5 Å ns – μs Medium scale loop motions SSE formation < 1 Å 0.01 ps 0.1 ps 1 ps Local: bond stretching angle bending methyl rotation
Amplitude Timescale Type of motion
Periodic (harmonic) Random (stochastic)
67
68
(Vijay Pande, Stanford University)
http://folding.stanford.edu/
69
70
1 10 100 1000 10000 100000 1 10 100 1000 10000 100000
experimental measurement (nanoseconds) Predicted folding tim e ( nanoseconds) PPA alpha helix beta hairpin villin
villin: Raleigh, et al, SUNY, Stony Brook BBAW: Gruebele, et al, UIUC beta hairpin: Eaton, et al, NIH alpha helix: Eaton, et al, NIH PPA: Gruebele, et al, UIUC BBAW
http://pande.stanford.edu/
71
Generate a large number
Select the correct, native-like fold Need good decoy structures Need a good energy function
72
73
1) Sequences of target proteins are made available to CASP participants in June-July of a CASP year
in the PDB, or even accessible 2) CASP participants have between 2 weeks and 2 months over the summer of a CASP year to generate up to 5 models for each of the target they are interested in. 3) Model structures are assessed against experimental structure 4) CASP participants meet in December to discuss results
74
28965 166 87 CASP6 22909 175 67 CASP5 5150 111 43 CASP4 1256 61 43 CASP3 947 72 42 CASP2 100 35 33 CASP1 # of 3D models # of predictors # of Targets Experiment
75
Three categories at CASP
CASP dynamics:
Venclovas, Zemla, Fidelis, Moult. Assessment of progress over the CASP experiments. Proteins, 53:585-595 (2003)
76
manner and select a native-like conformation using a good discrimination function
are not fooled by non-native conformations (or “decoys”)
77
sample conformational space such that native-like conformations are found astronomically large number of conformations 5 states/100 residues = 5100 = 1070 select hard to design functions that are not fooled by non-native conformations (“decoys”)
78
energy
79
x(t+Δt) = x(t) + v(t)Δt + [4a(t) – a(t-Δt)] Δt2/6 v(t+Δt) = v(t) + [2a(t+Δt)+5a(t)-a(t-Δt)] Δt/6 Ukinetic = ½ Σ mivi(t)2 = ½ n KBT
new position
new velocity
acceleration acceleration
n is number of coordinates (not atoms)
80
coordinates; reaching a deep minimum is not trivial
energy value energy number of steps deep minimum starting conformation steepest descent conjugate gradient energy number of steps give up converge RMSD
81
more moves are accepted initially and then cooling)
⎟ ⎠ ⎞ ⎜ ⎝ ⎛ − ∝ kT ΔE exp P
82
conformations
83
enumerate all possible conformations view entire space (perfect partition function) computationally intractable: 5 states/100 residues = 5100 = 1070 possible conformations select must use discrete state models to minimise number of conformations explored
84
database of experimentally determined conformations; common parametres include pairwise atomic distances and amino acid burial/exposure.
85
several native-like structures
the score/energy, the lower the RMSD)
86
IVGGYTCAANSIPYQ VSLNSGSHFCGGSLI NSQWVVSAAHCYKSR IQVRLGEHNIDVLEG NEQFINAAKIITHPN FNGNTL...
http://bioinf.cs.ucl.ac.uk
87
* * * * * * * * * *
* * * * * * * * * * * *
targets
* *
(Figure idea by Steve Brenner.)