SLIDE 1
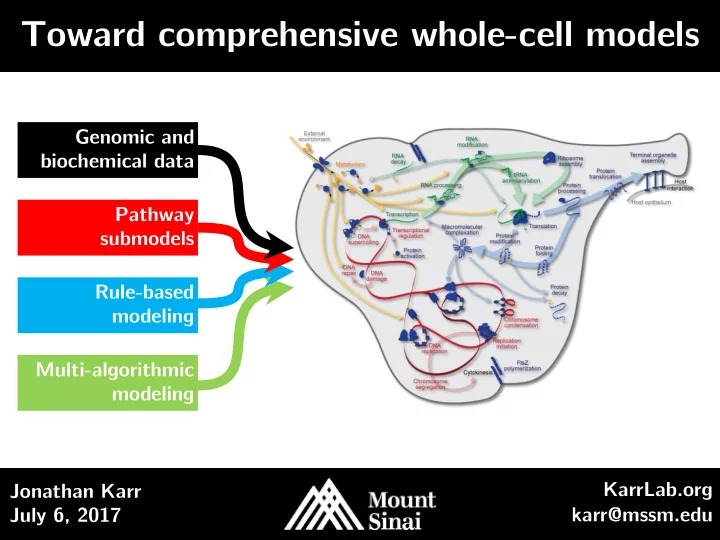
Toward comprehensive whole-cell models
KarrLab.org karr@mssm.edu Jonathan Karr July 6, 2017 Genomic and biochemical data Pathway submodels Rule-based modeling Multi-algorithmic modeling
SLIDE 2 Outline
Introduction to whole-cell (WC) modeling
- What is a WC model?
- Motivation
- Challenges
- Feasibility
Methodology
- Data aggregation
- Data organization
- Hybrid simulation
New tools to accelerate WC modeling
- Data aggregation
- Model representation
- Parallel simulation
SLIDE 3 Outline
Introduction to whole-cell (WC) modeling
- What is a WC model?
- Motivation
- Challenges
- Feasibility
Methodology
- Data aggregation
- Data organization
- Hybrid simulation
New tools to accelerate WC modeling
- Data aggregation
- Model representation
- Parallel simulation
SLIDE 4 Features of whole-cell (WC) models
Karr et al., 2015
Whole organism Dynamic Whole genome including each gene Stochastic Whole cell cycle Accurate Species-specific Mechanistic
AGTC
SLIDE 5 Outline
Introduction to whole-cell (WC) modeling
- What is a WC model?
- Motivation
- Challenges
- Feasibility
Methodology
- Data aggregation
- Data organization
- Hybrid simulation
New tools to accelerate WC modeling
- Data aggregation
- Model representation
- Parallel simulation
SLIDE 6
Genome design requires WC models
Tissue engineering Biosensors Biofactories
SLIDE 7
Example: drug biosynthesis
SLIDE 8
Example: drug biosynthesis
SLIDE 9
Example: drug biosynthesis
SLIDE 10
Example: drug biosynthesis
SLIDE 11
Example: drug biosynthesis
SLIDE 12
Example: drug biosynthesis
SLIDE 13
Example: drug biosynthesis
SLIDE 14
Personalized medicine requires WC models
SLIDE 15 Outline
Introduction to whole-cell (WC) modeling
- What is a WC model?
- Motivation
- Challenges
- Feasibility
Methodology
- Data aggregation
- Data organization
- Hybrid simulation
New tools to accelerate WC modeling
- Data aggregation
- Model representation
- Parallel simulation
SLIDE 16
WC models are a grand challenge
SLIDE 17 Time Length
Replication Growth Transcription Metabolism
Challenge: multiple time and length scales
SLIDE 18 Single-cell variation Microscopy Transcription RNA-seq Protein expression Mass-spec, Western blot
Challenge: heterogeneous data
SLIDE 19
Challenge: sparse data
SLIDE 20 Metabolic Signaling Transcriptional regulatory
Challenge: heterogeneous granularity
SLIDE 21 Outline
Introduction to whole-cell (WC) modeling
- What is a WC model?
- Motivation
- Challenges
- Feasibility
Methodology
- Data aggregation
- Data organization
- Hybrid simulation
New tools to accelerate WC modeling
- Data aggregation
- Model representation
- Parallel simulation
SLIDE 22
WC modeling is now feasible
Genomic and biochemical data Pathway submodels Rule-based modeling Multi-algorithmic modeling
SLIDE 23
Extensive molecular data is available
SLIDE 24 Numerous predictors are available
- miRNA targets: TargetScan
- Operons: OperonPredictor
- Protein half-lives: N-end rule
- Protein localization: PSORT
- Signal sequences: SignalP
- Transcription start site: Promoter
SLIDE 25
Numerous databases are available
SLIDE 26
Model design tools are available
MetaFlux
SLIDE 27
Model languages are available
SLIDE 28
Numerous pathway models are available
SLIDE 29 Numerous simulators are available
Uptake FBA Composition Metabolism FBA Composition Transcription Stochastic binding Gene expression Translation Stochastic binding Gene expression Replication Chemical kinetics DNA sequence
SLIDE 30
Testing tools are available
PRISM
SLIDE 31 Numerous other tools
- Automated model construction
- Model refinement
- Parallel simulation
- Calibration
- Analysis and visualization
- …
SLIDE 32 Outline
Introduction to whole-cell (WC) modeling
- What is a WC model?
- Motivation
- Challenges
- Feasibility
Methodology
- Data aggregation
- Data organization
- Hybrid simulation
New tools to accelerate WC modeling
- Data aggregation
- Model representation
- Parallel simulation
SLIDE 33 Pathway modeling workflow
- 1. Choose a system to model
- 2. Determine the scope and granularity of the model
- 3. Determine the mathematical representation of the
model
- 4. Reconstruct the species, reactions, rate laws, and
rate parameters from the literature
- 5. Debug and calibrate the model by comparison to
data
- 6. Test the model by comparison to independent data
SLIDE 34
Detail Scope
ODE Shuler, 1970’s FBA Palsson, 1990’s Boolean Bolouri, 2000’s Gillespie Luthey-Schulten, 2011 PDE
WC model
Predictive modeling methodologies
SLIDE 35 Scaling pathway modeling to whole-cells
– Accelerate data aggregation through automation – Organize input data using pathway/genome databases
- Build models collaboratively using web-based tools
– Define the semantic meaning of every model component – Track every assumption and data source
– Explicitly describe the data used to build models – Describe models in terms of rules
- Describe and simulate hybrid models
SLIDE 36
Scaling pathway modeling to whole-cells
Genomics, bioinformatics ↔ Mechanistic modeling Pathway/genome databases ↔ Model design tools Polymers, sequences ↔ Rule-based modeling Stochastic modeling ↔ Steady-state modeling (FBA) Numerical simulation ↔ Big data analytics Model design tools ↔ Collaboration tools
SLIDE 37
WC modeling workflow
SLIDE 38 Aggregate data
Fraser et al., 1995; Kühner et al., 2009; Lluch-Senar et al., 2013; Maier et al., 2013; Yus et al. 2012
Proteome Mass-spectrometry Transcriptome RNA-seq Epigenome Meth-seq Genome DNA-seq Metabolome Mass-spectrometry
SLIDE 39 Karr et al., 2013
Organize input data
SLIDE 40 Free Bound Promoter Bound Active
- 1. Update RNA polymerase states
- 3. Bind RNA polymerase
- 2. Calculate promoter affinities
- 4. Elongate and terminate transcripts
AUGAUCCGUCUCUAAUGUCUAC UTCAACGUGAGGUAAUAAAGUC UCCACGAUGCUACUGUAUC GCCUCAUACUGCGGAU UUACGUAUCAGUGAUCAGUACU Sequence Transcript
HcrA Spx Fur GntR LuxR glpF dnaJ dnaK gntR trxB polC
Design submodels
SLIDE 41 Design pathway submodels
Uptake FBA Composition Metabolism FBA Composition Transcription Stochastic events Gene expression Translation Stochastic events Gene expression Replication Chemical kinetics DNA sequence
SLIDE 42 Uptake FBA Composition Metabolism FBA Composition Transcription Stochastic events Gene expression Translation Stochastic events Gene expression Replication Chemical kinetics DNA sequence
Submodels States
Mass, shape Metabolite, RNA, protein counts Mammalian host Transcript, polypeptide sequences DNA polymerization, proteins, modifications FtsZ ring
Combine submodels
SLIDE 43 1 s
Uptake Metabolism Transcription Translation Replication
Cell states Cell states
Uptake Metabolism Transcription Translation Replication
Cell states
Uptake Metabolism Transcription Translation Replication
Concurrently integrate submodels
SLIDE 44
Calibrate model
1.Estimate individual parameters 2.Generate reduced models of individual pathways and to calibrate individual pathways 3.Refine joint parameter values using full models
SLIDE 45 Verify model against known biology
Matches training data
Cell mass, volume Biomass composition RNA, protein expression, half-lives Superhelicity
Matches published data
Metabolite concentrations DNA-bound protein density Gene essentiality
Matches theory
Mass conservation Central dogma Cell theory Evolution
No obvious errors
Plot model predictions Manually inspect data Compare to known biology
SLIDE 46 Verify model against known biology
Matches training data
Cell mass, volume Biomass composition RNA, protein expression, half-lives Superhelicity
Matches published data
Metabolite concentrations DNA-bound protein density Gene essentiality
Matches theory
Mass conservation Central dogma Cell theory Evolution
No obvious errors
Plot model predictions Manually inspect data Compare to known biology
SLIDE 47 Verify model against known biology
Matches training data
Cell mass, volume Biomass composition RNA, protein expression, half-lives Superhelicity
Matches published data
Metabolite concentrations DNA-bound protein density Gene essentiality
Matches theory
Mass conservation Central dogma Cell theory Evolution
No obvious errors
Plot model predictions Manually inspect data Compare to known biology
SLIDE 48 Verify model against known biology
Matches training data
Cell mass, volume Biomass composition RNA, protein expression, half-lives Superhelicity
Matches published data
Metabolite concentrations DNA-bound protein density Gene essentiality
Matches theory
Mass conservation Central dogma Cell theory Evolution
No obvious errors
Plot model predictions Manually inspect data Compare to known biology
SLIDE 49 Verify model against known biology
Matches training data
Cell mass, volume Biomass composition RNA, protein expression, half-lives Superhelicity
Matches published data
Metabolite concentrations DNA-bound protein density Gene essentiality
Matches theory
Mass conservation Central dogma Cell theory Evolution
No obvious errors
Plot model predictions Manually inspect data Compare to known biology
SLIDE 50 Verify model against known biology
Matches training data
Cell mass, volume Biomass composition RNA, protein expression, half-lives Superhelicity
Matches published data
Metabolite concentrations DNA-bound protein density Gene essentiality
Matches theory
Mass conservation Central dogma Cell theory Evolution
No obvious errors
Plot model predictions Manually inspect data Compare to known biology
SLIDE 51 Verify model against known biology
Matches training data
Cell mass, volume Biomass composition RNA, protein expression, half-lives Superhelicity
Matches published data
Metabolite concentrations DNA-bound protein density Gene essentiality
Matches theory
Mass conservation Central dogma Cell theory Evolution
No obvious errors
Plot model predictions Manually inspect data Compare to known biology
SLIDE 52 Verify model against known biology
Matches training data
Cell mass, volume Biomass composition RNA, protein expression, half-lives Superhelicity
Matches published data
Metabolite concentrations DNA-bound protein density Gene essentiality
Matches theory
Mass conservation Central dogma Cell theory Evolution
No obvious errors
Plot model predictions Manually inspect data Compare to known biology
SLIDE 53 Verify model against known biology
Matches training data
Cell mass, volume Biomass composition RNA, protein expression, half-lives Superhelicity
Matches published data
Metabolite concentrations DNA-bound protein density Gene essentiality
Matches theory
Mass conservation Central dogma Cell theory Evolution
No obvious errors
Plot model predictions Manually inspect data Compare to known biology
SLIDE 54 Verify model against known biology
Matches training data
Cell mass, volume Biomass composition RNA, protein expression, half-lives Superhelicity
Matches published data
Metabolite concentrations DNA-bound protein density Gene essentiality
Matches theory
Mass conservation Central dogma Cell theory Evolution
No obvious errors
Plot model predictions Manually inspect data Compare to known biology
SLIDE 55 Verify model against known biology
Matches training data
Cell mass, volume Biomass composition RNA, protein expression, half-lives Superhelicity
Matches published data
Metabolite concentrations DNA-bound protein density Gene essentiality
Matches theory
Mass conservation Central dogma Cell theory Evolution
No obvious errors
Plot model predictions Manually inspect data Compare to known biology
SLIDE 56
Genome 4700 kb 580 kb Genes 4461 525 Size 2 μm × 0.5 μm 0.2-0.3 μm
Mycoplasmas are small in size and complexity
SLIDE 57 Karr et al., 2012
Model includes 28 pathway submodels
SLIDE 58 Karr et al., 2012
Submodels are integrated through 16 common states
SLIDE 59 Model represents 76% of genes
1 10 100
Genes Process
DNA RNA Protein Other
Condensation Segregation Damage Repair Replication Rep Init Trans Reg Degradation Modification Processing Transcription Aminoacylation Complexation Activation Degradation Folding Modification Processing I Processing II Translocation Ribosome Term Org Translation Shape Cytokinesis FtsZ Metabolism
SLIDE 60
SLIDE 61 v v
Karr et al., 2012
Predict energy consumption
SLIDE 62
WC models predict ancestral similarity
SLIDE 63 Optimal gene expression
- M. genitalium
- M. mycoides
- M. pneumoniae
SLIDE 64
Optimal architecture retains robustness
Optimal gene expression retains robustness
SLIDE 65 Purcell et al., 2013
WC models can inform synthetic designs
SLIDE 66 Kazakiewicz et al., 2015
WC models can reposition antibiotics
SLIDE 67 Outline
Introduction to whole-cell (WC) modeling
- What is a WC model?
- Motivation
- Challenges
- Feasibility
Methodology
- Data aggregation
- Data organization
- Hybrid simulation
New tools to accelerate WC modeling
- Data aggregation
- Model representation
- Parallel simulation
SLIDE 68 Current limitations
- M. genitalium model is limited and inaccurate
- Ignores several pathways
- Mispredicts the growth rates of many single gene disruptions
Methods are not rigorous
- Data selection
- Multi-algorithm simulation
- Parameter estimation
- Verification
Methods are time-consuming
- Data aggregation
- Model design
- Model verification
Hard to understand, reuse, reproduce
SLIDE 69
Technology development goals
Scale to more complex models Accelerate WC model building and simulation Enable more researchers to engage in WC modeling Apply WC modeling to bioengineering and medicine
SLIDE 70
WC modeling process
SLIDE 71 Accelerate data aggregation
- Chaperones
- Complex composition
- DNA binding sites
- DNA footprints
- DNA methylation
- DNA sequence
- Gene-drug interactions
- Genome annotation
- Growth rates
- Metabolite concentrations
- Protein cofactors
- Protein expression
- Protein half-lives
- Protein localization
- Protein modification
- RNA editing
- RNA expression
- RNA half-lives
- RNA modification
- RNA maturation
- Reaction fluxes
- Reaction kinetics
- Reaction stoichiometries
- Signaling pathways
- DNA mutations
SLIDE 72 Accelerate data aggregation
- Help modelers quickly get relevant data for a
model
- Enable modelers to aggregate data
collaboratively
- Record the provenance of all data
Yosef Roth
SLIDE 73 Accelerate data aggregation
- 1. Merge data from as many sources as
feasible
- 2. Find most relevant data for the model
- Species: taxonomic distance
- Environment: temperature, pH, media
- 3. Normalize data
- 4. Calculate weighted consensus of the
relevant data
SLIDE 74
Accelerate data aggregation
SLIDE 75 Accelerate data aggregation
- Metabolite concentrations: ECMDB, YMDB
- RNA expression: Array Express
- Protein expression: PaxDB
- Protein complexes: CORUM
- Protein localization: prediction
- Protein-DNA interactions: DBD, DBTBS
- Reaction kinetics: SABIO-RK
SLIDE 76
Organize data for model building
SLIDE 77
Organize data for model building
SLIDE 78
Systemize model descriptions
John Sekar
SLIDE 79 Systemize model descriptions
Initiation
Dna(pos=sample(d.tss.pos)) + RnaPol -> Dna(pos=.).RnaPol.Rna(seq=‘’) algorithm: SSA rate law: constants: kcat value = d.tss.rate refs: [10.1093/bioinformatics/btw598, …] units = 1/s
Elongation
Dna(pos=<i>).RnaPol.Rna(seq=<j>) + RevComp(DsDna(pos=<i>)) -> DsDna(pos=<i+1>).RnaPol.Rna(seq=<j>+RevComp(DsDna(pos=<i>))) x-ref: EC: x.x.x.x
84
Rule-based modeling Genomic data Bioinformatic calculations Annotation, provenance Multi-algorithmic modeling
John Sekar
James Faeder, U Pitt
SLIDE 80 Language enables compact model descriptions
Initiation Elongation Termination SBML 1 per RNA 335 1 per base ~500k 1 per RNA 335 BioNetGen 1 per RNA 335 1 per RNA 335 1 per RNA 335 WC-Lang 1 1 1 1 1 1
85
SLIDE 81
Provenance tracking
SLIDE 82 Submodel design
WC-ML, SBML, CellML
SLIDE 83 Systemize simulation
Goldberg et al., 2016
Arthur Goldberg
SLIDE 84
High-performance simulator
SLIDE 85
Simulation results database
SLIDE 86
Visual analysis of simulation results
SLIDE 87 Future work
- Online platform for collaborative model
design
- Parallel, rule-based simulator
- Scalable methods for calibrating and
validating large models
- Community standard for verifying WC
models
SLIDE 88 Karr Lab overview
Technology development Modeling language
- Programmatic
- Rule- and sequence-based
- Multi-algorithmic
Parallel simulation
- Reusable
- Multi-algorithmic
- Parallel discrete event
simulation
Pilot models
- M. pneumoniae
- Expand scope
- Improve accuracy
- Drive genome design
Stem cells
- Personalized models
- Precision medicine
WC- Rules
SLIDE 89
Summary
Genomic and biochemical data Pathway submodels Rule-based modeling Multi-algorithmic modeling
SLIDE 90 Methods development James Faeder, U Pitt
Veronica Llorens, CRG Maria Lluch-Senar, CRG Samuel Miravet, CRG Luis Serrano, CRG
Pablo Meyer, IBM
Acknowledgements
John Sekar Yosef Roth Roger Rodriguez Arthur Goldberg Yin Hoon Chew Balazs Szigeti
SLIDE 91
Summary
Genomic and biochemical data Pathway submodels Rule-based modeling Multi-algorithmic modeling