
MOL2NET, 2018, 4, http://sciforum
MDPI
MOL2NET
Development and Ch Coated Con
Sateesh Kumar Vemula1*, R
1Department of Pharmaceutics, M 2Department of Pharmaceutics, A 3Sri Shivani College 4Chaitanya College of Pharmac
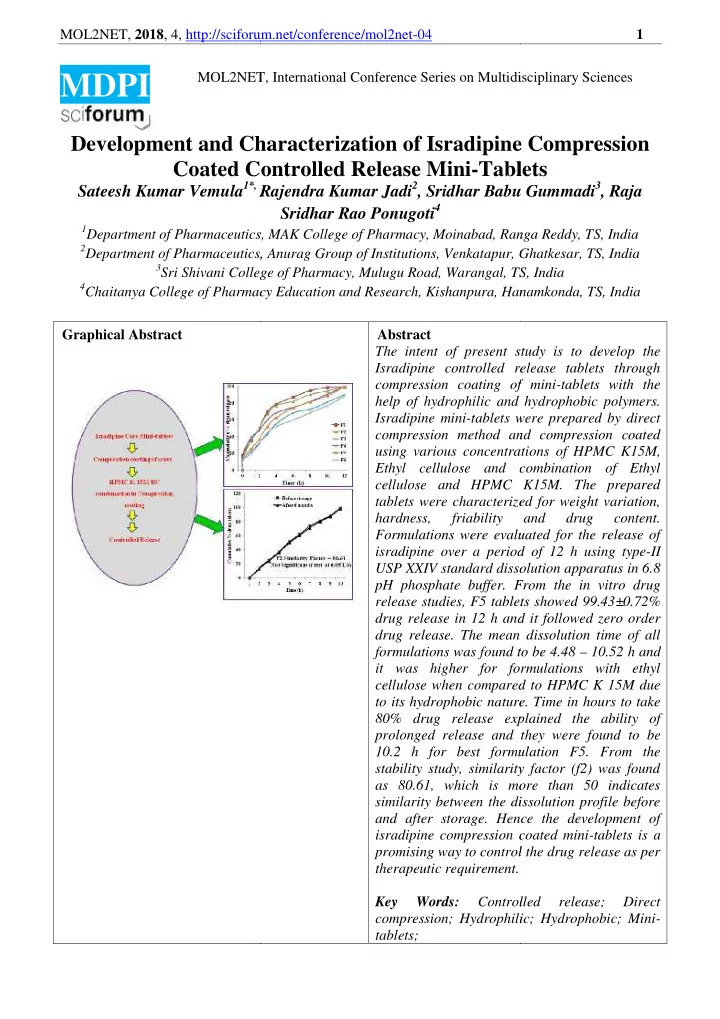
Graphical Abstract
- rum.net/conference/mol2net-04
ET, International Conference Series on Multidisc
Characterization of Isradipine Controlled Release Mini-Table
*, Rajendra Kumar Jadi2, Sridhar Babu
Sridhar Rao Ponugoti4
ics, MAK College of Pharmacy, Moinabad, Ran cs, Anurag Group of Institutions, Venkatapur, G
- llege of Pharmacy, Mulugu Road, Warangal, TS
acy Education and Research, Kishanpura, Hanam Abstract The intent of present st Isradipine controlled re compression coating of help of hydrophilic and Isradipine mini-tablets w compression method an using various concentrat Ethyl cellulose and c cellulose and HPMC tablets were characterize hardness, friability Formulations were evaluat isradipine over a period USP XXIV standard dissol pH phosphate buffer. From release studies, F5 tablet drug release in 12 h and drug release. The mean formulations was found to it was higher for form cellulose when compared to its hydrophobic nature 80% drug release expl prolonged release and t 10.2 h for best formul stability study, similarity as 80.61, which is more similarity between the dissol and after storage. Henc isradipine compression c promising way to control therapeutic requirement. Key Words: Controll compression; Hydrophili tablets; 1 idisciplinary Sciences
ne Compression Tablets
bu Gummadi3, Raja
anga Reddy, TS, India apur, Ghatkesar, TS, India , TS, India anamkonda, TS, India study is to develop the release tablets through
- f mini-tablets with the
and hydrophobic polymers. s were prepared by direct and compression coated rations of HPMC K15M, and combination of Ethyl
- K15M. The prepared
rized for weight variation, and drug content. aluated for the release of riod of 12 h using type-II ssolution apparatus in 6.8
- r. From the in vitro drug
ablets showed 99.43±0.72% and it followed zero order an dissolution time of all
- und to be 4.48 – 10.52 h and
formulations with ethyl pared to HPMC K 15M due
- ure. Time in hours to take
xplained the ability of and they were found to be
- rmulation F5. From the
ity factor (f2) was found more than 50 indicates dissolution profile before ence the development of
- n coated mini-tablets is a
rol the drug release as per nt. rolled release; Direct drophilic; Hydrophobic; Mini-