
SLIDE 1
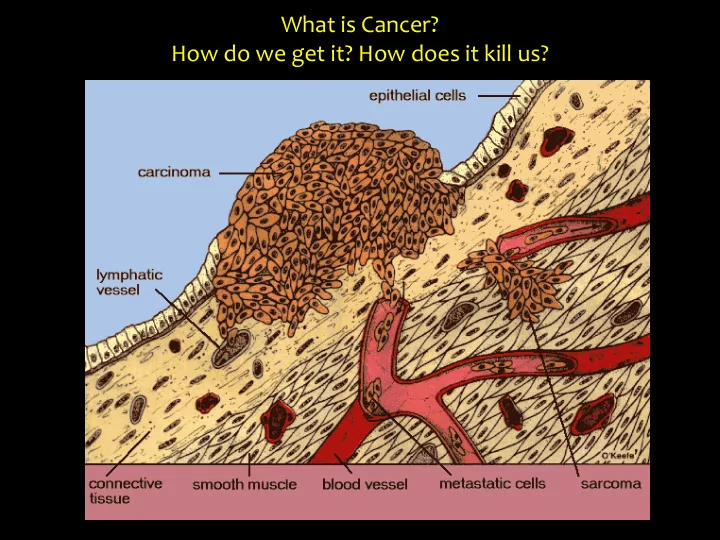
How do we get it? How does it kill us? Two distinct cancer research - - PowerPoint PPT Presentation
What is Cancer? How do we get it? How does it kill us? Two distinct cancer research themes: 1. tumor onset (how does it all start? Is it really just a question of bad luck?) 2. metastasis and phenotypic plasticity (what has personalized
Two distinct cancer research themes:
(how does it all start? Is it really just a question of bad luck?)
(what has personalized medicine to do with that?)
Colorectal Cancer: the second deadliest
sequential clonal expansions (Darwin docet!)
2nd hit (gene B mutation) 1st hit (gene A mutation) 3rd hit (gene C mutation)
intestinal epithelial cell ACF adenoma
p53
carcinoma
The Adenoma-Carcinoma Sequence in Colorectal Cancer: a genetic paradigm for tumor initiation, progression, and metastasis
APC K-RAS SMAD2/4
What about the rate-limiting, initiating (APC) mutation event? Is it just a ‘random’ event (bad luck, shit happens), or do extrinsic (environmental) factors favor its occurrence?
Cancer: the role of “bad luck” vs. environmental risk factors
Cancer: the role of “bad luck” vs. environmental risk factors
Colorectal Cancer: the second deadliest, highest stem cell division rate (1012 over 60 yrs), and yet largely due to environmental factors.
Cancer: the role of “bad luck” vs. environmental risk factors
Why do we get colon cancer? 4 main etiological factors:
It’s all in the stem cell niche!
intestinal epithelial cell ACF adenoma
p53
carcinoma
APC K-RAS SMAD2/4
stem cell niche alterations
(i.e. expansion of cell targets for tumor initiation and progression)
metaplasia
(stem cell reprogramming)
Western-style diet Aging IBD Predisposition
Why do we get colon cancer? 4 main etiological factors:
It’s all in the stem cell niche!
NWD1: - 25% of C57BL/6J mice develop colon tumors (n= 1-2);
→ First and only model of dietary induced sporadic CRC NWD2: Increased Vit D3 and Ca2+ rescue the tumorigenic effect
0% 100% 200% 300% 400% 500% Fat Vit D3 Ca2+ Fiber Methyl donors % of control diet AIN76A NWD1 NWD2
1 2 3 4 5 6 7 8 9 10
3% DSS H20
SACRIFICE
Lgr5-EGFP
1 2 3 4 5 6 7 8 9 10
H20
SACRIFICE
Lgr5-EGFP
cKit+ Paneth-like cells (in colon) LyscreERT2
(J. Van Es, H. Clevers)
cKitcreERT2
(D. Saur)
Lgr5EGFP-IRES-creERT2
(N. Barker, H. Clevers)
GFP
Control 3% DSS
GFP GFP/RFP GFP/RFP
GFP/YFP GFP/YFP GFP
Control diet NWD1 diet
GFP GFP Ki67 GFP Ki67
→ fewer Lgr5+ stem cells → Lgr5+ stem cell lineage tracing is inhibited
→ Lgr5+ stem cells cease to proliferate
NWD1 inhibits proliferation, lineage tracing, and tumor-initiating capacity
The western-style NWD1 diet (low in vitamin D) represses the stem cell function of Lgr5+ cells and their capacity to underlie cell turnover in the small an large intestine. Are there other stem cells involved? Or are the dietary effects mediated by the niche?
small intestine
crypts 500 crypts/ well
Matrigel + growth factors
0h 5h 1day 5 days
AIN76A NWD1 NWD2 5x 20x 10x
20 40 60 80 100 120 140 160 180 200 AIN76A NWD1 NWD2 number of organoids
→ notwithstanding the fact that the Western-style diet suppresses stemness of Lgr5+ cells, mini-gut organoid forming capacity is increased. Hence, a distinct stem cell type is activated upon Western-style diet. Intestinal stemness is increased by Western-style diet
5 day old organoids
1000 Lgr5+ CBCs from mouse A 1000 Paneth cells from mouse B Plate out in matrigel for ‘mini-gut’ assay 30’ co-incubation at 37°C
modification of sorted lineages
(e.g. control diet)
(e.g. NWD1 diet)
Single cell digest of intestinal mucosa FACSort stem (Lgr5+) and niche (Paneth) cells separately
Functional analysis of stem vs. niche effects: the organoid reconstitution assay
Lgr5 cells Paneth cells
Western style diet
Lgr5+ stem cells inactivation
Homeostasis
Paneth cells provide niche support to Lgr5+ stem cells. +4 Lgr5+ Lyz+Paneth
Inflammation
Depletion of Lgr5+ stem cells
Regeneration
Paneth cells acquire stem cell properties and repopulate the intestinal epithelium.
Lineage tracing Lineage tracing
Regeneration
Niche reinforcement by Paneth cells, quiescent stem cell (+4) activation. +4 Lyz+Paneth
Overall, colon cancer risk factors such as diet and inflammation profoundly affect both niche and stem cells resulting in improved regeneration upon tissue injury. The recruitment of new stem cell types and the overall increased proliferation rate ultimately results in an increased mutation rate and cancer risk.
Two distinct cancer research themes:
(how does it all start? Is it really just a question of bad luck?)
(what has personalized medicine to do with that?)
20 30 40 50 60 70
Age (years) (epi)genetic mutation load
Tumor Evolution at the Primary Site (Darwin docet: sequential clonal expansion)
2nd hit 1st hit 3rd hit 2nd hit
3rd hit
EMT
surgery
Dissemination (CTCs & DTCs)
genetic hit (mutation) epigenetic hit (red: EMT; yellow: MET)
Distant Site Metastases
1st hit
The current ‘hype’ on personalized cancer therapy is misplaced and unjustified
‘a large majority of driver gene mutations are common to all metastases’ ‘the driver gene mutations that were not shared by all metastases are unlikely to have functional consequences’ 2002 2018
Tumor cells are capable of contributing to normal development and generate phenotypically normal chimeric organism with tumor cells present in most adult tissues
(mouse teratocarcinoma cells: Illmensee & Mintz, 1976; chicken embryonic cells infected with Rous sarcoma virus: Rous, 1979; Dolberg & Bissell, 1984; Stoker, Hatier, & Bissell, 1990).
Som
earl rly (70’s and 80’s) exp xperimental evid viden ence th that t th thin ings gs are not
imple e as th that:
Mina Bissel, PhD:
phenotype is dominant over genotype!!
Notably, when cultured in vitro, these embryonic cells rapidly reveal pronounced tumorigenic features and undergo neoplastic transformation. Hence, the embryonic environment seems to attenuate oncogenesis possibly by inducing differentiation and suppressing cancer stemness.
Type, density and location of tumor-infiltrating immune cells in large cohorts of human colorectal cancers represent the strongest prognostic factors in terms of freedom from disease and overall survival at all stages of clinical disease (Galon et al., Science 2006).
Jerome Galone, PhD
The Overhyping of Precision Medicine
Science has a history of inflated promises when it comes to disease treatment. by Nathaniel Comfort, Dec. 16, 2016
www.theatlantic.com/health/archive/2016/12/the-peril-of-overhyping-precision-medicine/510326
….
Epithelial markers:
Mesenchymal markers:
Functional features:
Functional features:
Functional features:
adhesion
E/M markers:
Phenotypic Plasticity and Metastasis: conventional immortalized cell lines as experimental models
Common colon cancer cell lines encompass a CD44highEpCAMlow subpopulation with mesenchymal and highly motile and invasive features
Andrea Sacchetti
CD44highEpCAMlow colon cancer cells express
EMT markers and transcription factors (ZEB1)
CD44hiEpCAMlow =19% CD44hiEpCAMlow = 84%
EpCAM-FITC CD44-APC HCT116 - Sorted EpCAM low + 34 days
CD44highEpCAMlow cells have stem-like characteristics
CD44highEpCAMlow cells have high-metastatic capacity in vivo
HCT116 - Spleen injection
…and are resistant to chemotherapy
CD44hiEpCAMhigh CD44hiEpCAMlow
CD44hiEpCAMhigh CD44hiEpCAMlow
HCT116 – Top differentially expressed genes
EpCAM E-Cad ZEB1 VIM1 RSPO1
RNAseq analysis: functional and omics characterization
HCT116 and SW480: combined pathway analysis HCT116 and SW480: MDS analysis
CD44highEpCAMlow cells correlate with EMT signatures and the CMS4 subtype of colon cancer
HCT116 – chemotherapy
Post- treatment Pre- treatment Recurrence Post- treatment Pre- treatment Recurrence
SW480 – chemotherapy
CD44highEpCAMlow cells correlate with signatures of chemoresistance and have a worse clinical outcome
http://labriccardofodde.nl